近日,我中心唐海宝教授、黄育敏教授联合广西大学张积森教授于Plant Communications杂志发表题为NodeGWAS: Leveraging Graph Pangenomes for Sensitive and Accurate Association Analysis in Diverse Diploid and Polyploid Species的研究论文。该研究提出NodeGWAS方法——一个基于图形泛基因组的分析框架,通过节点开展关联分析。相较于传统全基因组关联分析方法,NodeGWAS在敏感性与可解释性之间实现了平衡,其在拟南芥和甘蔗中的应用证明了该方法在解析复杂性状方面的有效性。

全基因组关联分析(GWAS)是解析复杂性状遗传基础的核心手段。然而,传统基于线性参考基因组的方法在处理结构变异、高重复序列及多倍体物种时容易产生比对偏差,从而导致遗传信息丢失。尽管无参考基因组的k-merGWAS等方法能提升关联灵敏度,但仍存在冗余度高以及难以定位的问题。近年来,图形泛基因组展现出替代标准线性基因组的巨大潜力,但目前直接基于图形泛基因组拓扑结构进行变异检测和分型的研究仍较为有限,具有较大的应用前景。针对这一空白,研究团队开发了NodeGWAS 框架,旨在为遗传背景复杂或具有高结构变异多样性的物种提供兼具灵敏性与准确性的关联分析方案。
NodeGWAS方法直接基于图泛基因组拓朴结构,将图中的每个节点(Node)视为基本序列单元,通过精确构建群体的非冗余遗传特征矩阵开展关联分析。在图泛基因组中,各种变异,无论是SNP、INDEL还是复杂SV都会造成节点拓扑结构的变化。相比传统方法,NodeGWAS无需显式进行结构变异检测,从而避免了传统流程在复杂变异处理中所导致的信息损失及步骤冗余。NodeGWAS流程主要包括四个步骤:首先将个体重测序数据比对到图泛基因组,确定节点的存在/缺失状态;其次整合所有个体的节点信息构建群体水平的节点矩阵;随后结合群体结构与亲缘关系进行关联分析;最后将显著关联的节点映射回线性参考基因组进行下游分析。
在针对经典的拟南芥群体数据评估中,NodeGWAS 在完整捕获FT、FLC和VIN3等已知开花时间调控位点的基础上,额外发现了2个传统方法无法检测的性状关联区间信号。值得注意的是,NodeGWAS 在保持高灵敏度的同时,在假阳性控制方面也显著优于传统的无参考k-mer方法。此外,在根长性状分析中,NodeGWAS成功在EPR1基因附近发现了传统方法均无法检测到的新型显著位点。深入探究该位点的子图结构发现,信号源于一种多层嵌套的“巢式变异”(Nested Variation),这种复杂的拓扑结构往往会导致传统变异调用方法失效。
为了进一步展示NodeGWAS在多倍体中的适用性,研究将其应用于复杂多倍体甘蔗。在8个与糖含量相关的性状分析中,传统基于SNP的方法几乎无法显著关联,而k-merGWAS和NodeGWAS分别识别出4,751和544个位点。尽管k-mer方法在显著位点数量上占优,但两种方法之间的重叠较少,表明它们在信号检测上具有良好的互补性。NodeGWAS共检测出143个节点在超过一半性状中均显著关联,其中位于6号染色体约36 Mb区域的3个节点与4个糖含量相关性状显著关联。这三个节点被定位在基因Erufi.06G007100的启动子区域,该基因编码葡萄糖醛酸转移酶,且在甘蔗茎和叶中的表达水平与糖分积累呈现明确的相关性,提示其在糖分积累中的潜在调控作用。这些结果不仅表明了NodeGWAS在面对复杂多倍体物种时的有效性,也为甘蔗糖含量相关性状鉴定了新的候选基因。
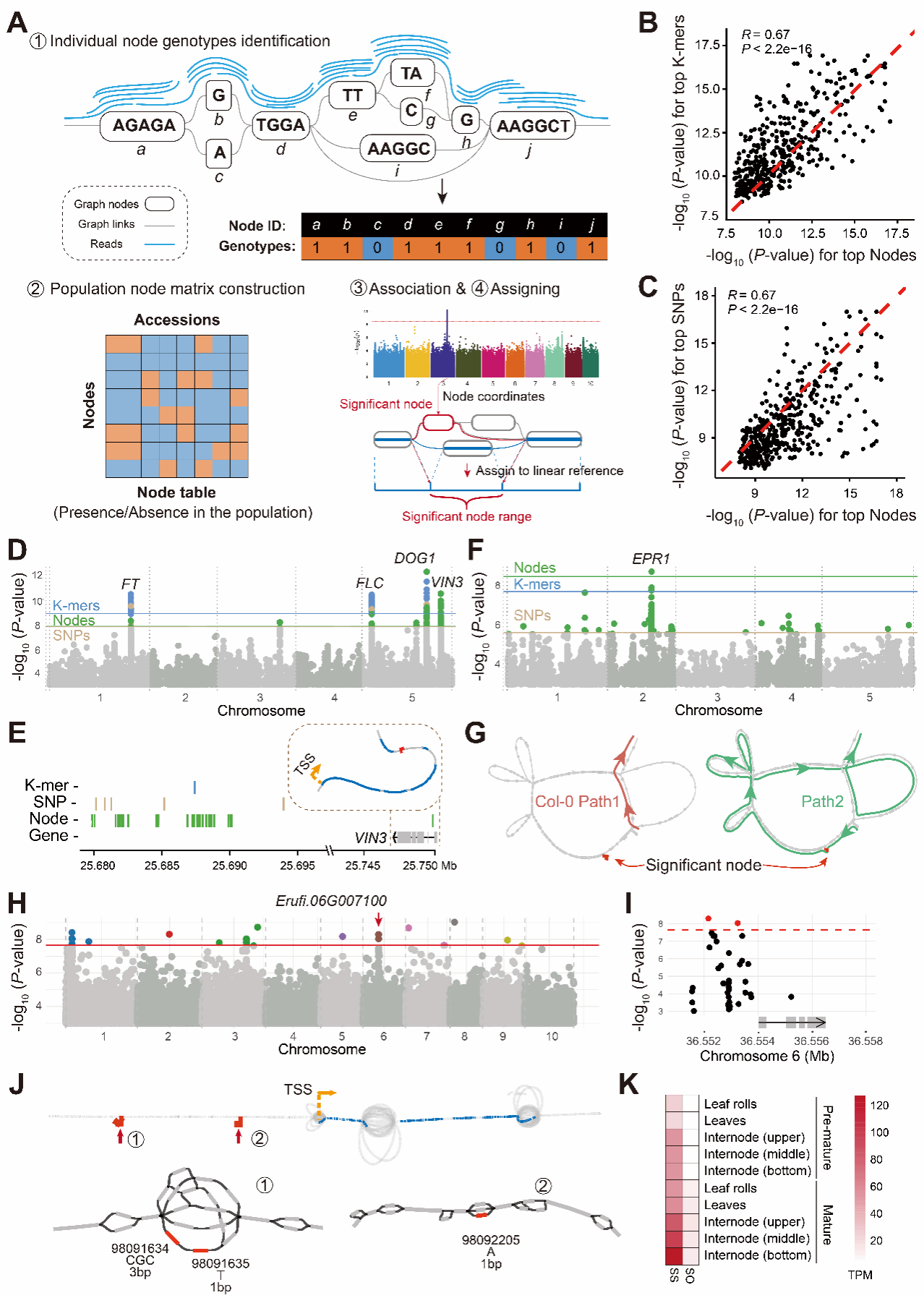
图1 NodeGWAS:流程、性能评估及其应用
总体而言,NodeGWAS 提供了一种基于图形泛基因组的创新性GWAS方法,聚焦非冗余序列变异的精确检测和关联,在降低结果冗余、提高假阳性控制能力以及增强对复杂基因组的适用性方面表现出显著优势,为二倍体和多倍体物种的关联分析提供了更加稳健且具有良好生物学解释性的解决方法。
研究团队
福建农林大学已毕业硕士生张以星(现广西大学亚热带农业生物资源保护与利用国家重点实验室科研助理)为论文的第一作者,福建农林大学唐海宝教授、黄育敏教授以及广西大学张积森教授为论文共同通讯作者。该研究得到国家重点研发计划、国家自然科学基金的资助。值得一提的是,该研究是基于团队前期构建的甘蔗多尺度图形泛基因组框架(Huang et al. Science, 2026)上的进一步扩展与演进,为复杂遗传背景下的关键性状解析提供了更为全面且互补的技术手段。
